Research
Nucleophilic substitution (SN) reactions account to the most essential and frequently applied transformations in chemistry. Nevertheless, they are typically associated inevitably with the generation of unfavourable waste amounts and are therefore restricted by a poor sustainability and a poor cost-efficiency.[1] To address these major challenges, we are committed towards the development of novel catalytic methods for SN-type bond formations as illustrated in Scheme 1 A.[2,3]

©Number1411/Shutterstock.com
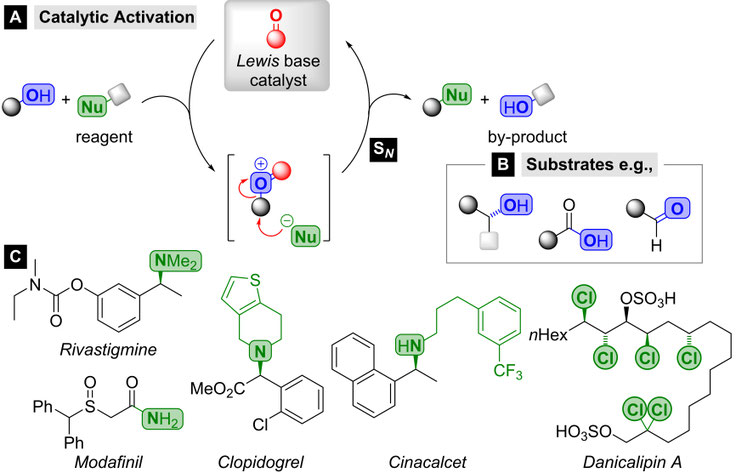
Scheme 1. A General concept for Lewis base catalyzed nucleophilic substitutions and C selected natural products and pharmaceuticals potentially accessible through SN-type transformations.
Thereby, mild reaction conditions and a good waste balance are enabled through the application of weakly electrophilic reagents such as benzoyl (BzCl),[4,6,7,10] acetyl[6,7] and cyanuric chloride (TCT) and phenyl chloroformate (Scheme 2).[9] These inexpensive commodity chemicals furnish only weakly acidic by-products, whereas conventional protocols are often restricted by the formation of strongly acidic hydrogen chloride. Notably, these non-classical agents only provide the desired substitution products in high levels of chemoselectivity in the presence of a suitable Lewis base catalysts. Indeed, we identified simple formamides such as 1-formylpyrrolidine as potent catalysts for the conversion of alcohols into alkyl chlorides by means of benzoyl chloride[4] and TCT in substoichiometric amounts for down to 34 mol% with respect to the starting alcohol[5] (Scheme 2 A).

Scheme 2. Lewis base catalyzed dehydroxyhalogenations of alcohols.[4-11] a. Prepared from corresponding alcohol (98-99% ee) under twofold inversion. Cbz = carbonyloxybenzyl TBDMS = tert-butyldimethylsilyl, MOM = methoxymethyl.
In the following, we introduced diethylcyclopropenone catalyzed chlorinations, brominations and iodinations of alcohols with benzoyl chloride and acetyl chloride, respectively, which are distinguished by low catalyst loadings (down to 1 mol%) and high turnover numbers up 100.[6] In addition, out of a rational screening of various sulfinyl compounds (2-methoxyphenyl) methyl sulfoxide emerged as potent catalyst to access benzylic chlorides.[7] Sulfoxide catalysis in regard to nucleopilic substitutions is mainly focused on benzylic substrates, whereas Pummerer rearrangement is the dominant reaction pathway in the case of less reactive aliphatic starting materials. Recently, phenyl chloro formate was discovered as safer substitute for the toxic gas phosgene in SN-reactions engaging alcohols.[9] Moreover, formamide catalysis also facilitated the synthesis of geminal dichlorides from aldehydes in high efficacy using phthaloyl chloride as stoichiometric promotor (Scheme 3).[10]

Scheme 3. Lewis base catalysis promoted synthesis of geminal dichlorides from aldehydes.[11] Phth = Phthaloyl.
Subsequently, formamide catalysis has been applied towards the activation of carboxylic acids (Scheme 4). Transformation into acid chlorides by means of TCT, which is the cheapest reagent for OH group activation (besides phosgene), allowed to construct C-O ester and C-N amide bonds in high yields and excellent scalability. Remarkably, even peptids are accessible virtually without epimerization, when phthaloyl protected amino acids are employed as starting materials.

Scheme 4. Formamide catalysis enabled preparation acid chlorides, esters and amides and selected examples.[12]
Eventually, FPyr facilitated the transformation of formic acid into CO by means of TCT (Scheme 5).[13] The formed CO was then harnessed ex situ for different transition metal catalyzed carbonylative transformations.

Scheme 4. Formamide catalysis promoted fransformation of formic acid into CO and application towards various carbonylative transformations.[13]
References
[1]: (a) Constable et al., Green Chem. 2007, 9, 411; (b) J. An, R. M. Denton, T. H. Lambert, E. D. Nacsa, Org. Biomol. Chem. 2014, 12, 2993–3003; (c) P. H. Huy, T. Hauch, I. Filbrich, Synlett 2016, 27, 2631-2636; (c) P. H. Huy, Eur. J. Org. Chem. 2020, 10-27.
[2]: Selected examples for Lewis-Base catalyzed halogenations of alcohols: (a) R. M. Denton, J. An, B. Adeniran, Chem. Commun. 2010, 46, 3025–3027; (b) R. M. Denton, J. An, B. Adeniran, A. J. Blake, W. Lewis, A. M. Poulton, J. Org. Chem. 2011, 76, 6749–6767; (c) C. M. Vanos, T. H. Lambert, Angew. Chem. Int. Ed. 2011, 50, 12222–12226; Angew. Chem. 2011, 123, 12430-12434; (d) H. A. van Kalkeren, S. H. A. M. Leenders, C. Rianne, A. Hommersom, F. P. J. T. Rutjes, F. L. van Delft, Chem. Eur. J. 2011, 17, 11290–11295; (e) C. Dai, J. M. R. Narayanam, C. R. J. Stephenson, Nature Chem. 2011, 3, 140-145; (f) T. V. Nguyen, A. Bekensir, Org. Lett. 2014, 16, 1720−1723; (g) L. Longwitz, S. Jopp, T. Werner, J. Org. Chem. 2019, 84, 7863-7870; (h) M. A. Hussein, T. V. Nguyen, Chem. Commun. 2019, 55, 7962-7965.
[3]: Catalytic Mitsunobu reactions: (a) T. Y. S. B. But, P. H. Toy, J. Am. Chem.
Soc. 2006, 128, 9636-9637. (b) O’Brien, C. J. PCT Int. Appl. WO2010/118042A2, 2010.
(c) D. Hirose, T. Taniguchi, H. Ishibashi, Angew. Chem. Int. Ed. 2013, 52, 4613–4617;
Angew. Chem. 2013, 125, 4711-4715; (d) J. A. Buonomo, C. C. Aldrich, Angew. Chem. Int. Ed. 2015, 54, 13041–13044; Angew.
Chem. 2015, 127, 13233-13236; (e) D. Hirose, M. Gazvoda, J. Kosmrlj, T. Taniguchi, Chem.
Sci. 2016, 7,
5148-5159; (f) D. Hirose, M. Gazvoda, J. Kosmrlj, T. Taniguchi, Org. Lett. 2016, 18, 4036−4039; (g) R. H.
Beddoe, K. G. Andrews, V. Magné, J. D. Cuthbertson, J. Saska, A. L. Shannon-Little, S. E. Shanahan, H. F. Sneddon, R. M. Denton, Science 2019, 365,
910-914.
[4]: P. H. Huy, S. Motsch, S. M. Kappler, Angew. Chem. 2016, 128, 10300-10304; Angew. Chem. Int. Ed. 2016, 55, 10145-10149.
[5]: P. H. Huy, I. Filbrich, Chem. Eur. J. 2018, 24, 7410-7416.
[6]: T. Stach, J. Dräger, P. H. Huy, Org. Lett. 2018, 20, 2980-2983.
[7]: S. Motsch, C. Schütz, P. H. Huy, Eur. J. Org. Chem. 2018, 4541-4547.
[9]: B. Zoller, T. Stach, P. H. Huy, ChemCatChem 2020, doi: 10.1002/cctc.202001175.
[10]: C. Kohlmeyer, S. Schäfer, P. H. Huy, G. Hilt, ACS Catalysis 2020, 10, 11567-11577.
[11]: P. H. Huy, Synthesis 2019, 51, 2474-2483.
[12]: P. H. Huy, C. Mbouhom, Chem. Sci. 2019, 10, 7399-7406.
[13]: B. Zoller, J. Zapp, P. H. Huy, Chem. Eur. J. 2020, 226, 9632-9638.

Prof. Dr. Peter Huy
Rostock University
Institute for Chemistry
Office 202
Albert-Einstein Straße 3a
18059 Rostock
Germany
phone +49 (0) 381 498 6440